Researchers have overlooked especially minuscule gene fragments that are critical to the assembly of cellular machinery and could help better trace the evolutionary history of plants, says a new study led by the University of Nebraska–Lincoln.
After analyzing the genomes of species ranging from algae to rice, Nebraska’s Chi Zhang, Huihui Yu and their colleagues identified more than 1,000 consequential gene fragments not accounted for in prior analyses.
Those fragments are known as exons: portions of genes that direct the production of amino acids, which themselves form the proteins responsible for all sorts of life-sustaining tasks. Correctly identifying and accounting for every exon in a gene, then, is essential to understanding how that gene influences the function or dysfunction of a cell.
Most exons contain many dozens or even hundreds of nucleotides, the chemical compounds — in DNA, adenine (A), cytosine (C), guanine (G) and thymine (T) — that pair up to form the rungs of DNA’s double-stranded ladder. Nucleotide sequences ultimately get transcribed into messenger RNA, which then ferries its assembly guide to a cell’s ribosome, where the corresponding protein gets manufactured.
But some genes also include exons that feature far fewer nucleotides.
“We found that there are many very small micro-exons that are shorter than 15 nucleotides,” said Zhang, professor of biological sciences at Nebraska. “Some exons may consist of only one nucleotide. It was a little surprising even to us.”

Their diminutive size makes micro-exons more difficult to identify, Zhang said, especially given that they reside between so-called introns, the portions of a gene that play no part in the assembly of a protein. As short as they are, though, micro-exons are no less important to constructing, and understanding, a functioning protein. And because every nucleotide in an exon is part of a three-nucleotide sequence that corresponds to a particular amino acid, overlooking even one can lead researchers to misconstrue the entire blueprint of a protein.
“If there is one inversion or one deletion or one shift of a nucleotide, it will create a completely different protein sequence,” Zhang said.
So the team set out to develop a computational tool that could better recognize and classify the micro-exons hiding among the introns. In a series of simulations, the tool outperformed two commonly used counterparts when it came to identifying micro-exons and avoiding false positives.
After comparing its results against the published RNA sequences of 10 species, the Nebraska team concluded that up to 81% of micro-exons in rice, and 65% in corn, have been overlooked. Some of the proteins built from the blueprints of those missing micro-exons are known for helping the crops adapt to environmental stresses by controlling the development of shoots, roots and flowers.
“We found that, across genomes, there are about 3% of expressed genes that have micro-exons,” Zhang said. “It’s not a small number. That can cause us to completely misunderstand a whole system.
“Our discovery shows that, contrary to the current understanding, micro-exons are actually much more prevalent.”
Realizing just how many micro-exons have gone overlooked also got the team wondering about their role in the evolution and divergence of plant species. But finding instances of similar or even identical exons across species is, again, much tougher when they’re micro, Zhang said.
“For example, if we have only one nucleotide, how can we compare that to others,” he said with a rueful laugh. “Even if we have 15 nucleotides, it’s still too short to conduct a sequence comparison.”
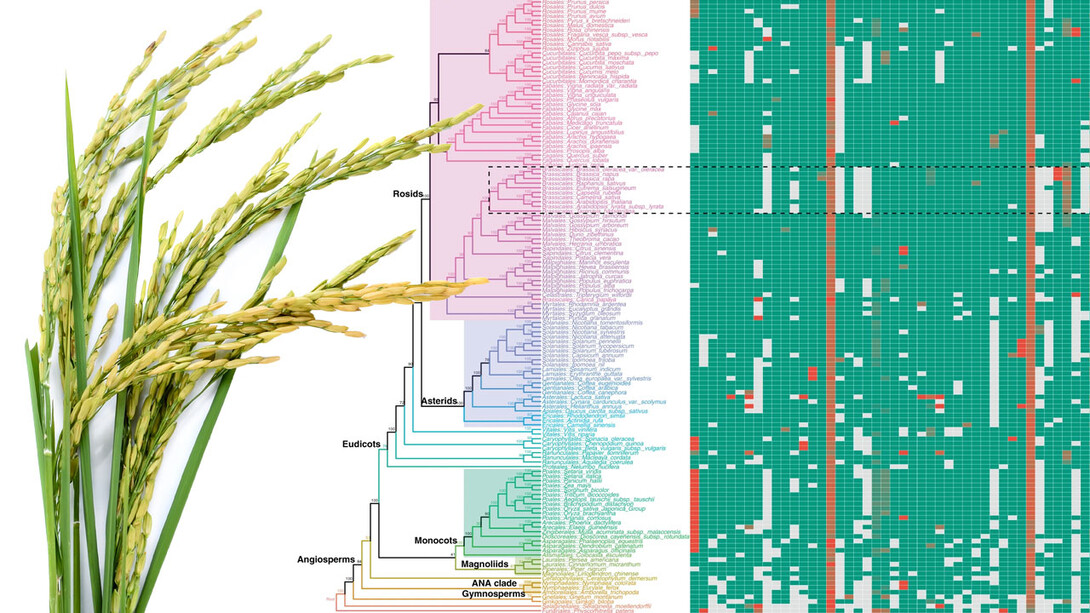
The team’s solution? Compile the 108 exon nucleotides that flank either side of a micro-exon, yielding a “micro-exon tag” that the researchers could then use to search for counterparts across the genomes of other species. When they did, the researchers found that 45 micro-exon clusters appeared in at least three of the nine terrestrial plant species they analyzed.
With those 45 clusters as a starting point, the team also developed a model for predicting the presence of micro-exons in a plant species. That model correctly predicted more than 90% of the micro-exons from the 45 clusters when put to the test across four of the plant species. So the team proceeded to apply its model to the genomes of 132 plants — about 400 terabytes worth of what Zhang called “real big data” — from 10 species.
The model predicted that 35 of the 45 micro-exon clusters are shared in flowering plants and moss but not green algae, suggesting that roughly 80% of modern micro-exons stretch back to the common ancestor of all land-based plants. In fact, Zhang’s team discovered that an evolutionary tree based on micro-exons alone would resemble the trees commonly accepted by evolutionary biologists. And that, Zhang said, makes micro-exons a genetic equivalent of “amber fossils, trapped in time.”
“These micro-exon clusters are a good set of markers for studying evolution, because they are conserved (across species and time),” he said. “So this is a very important discovery. In the future, when more genome sequences are assembled, we can use micro-exon markers to review the details of plant evolution.”
Zhang said micro-exons also offer a prime opportunity to study a phenomenon that’s essential in transforming them from fragments of DNA into the structure-granting joints of proteins. That phenomenon, splicing, involves removing the introns from between the exons of RNA and linking those exons together — essentially, cutting genetic chaff from wheat. Usually, splicing occurs before the addition of a cap and tail that keep the resulting messenger RNA from degrading on its way to the protein-manufacturing ribosome.
But Zhang’s team found that micro-exons flip the transcript. Instead of undergoing the splicing first, the messenger RNAs housing micro-exons often don their caps and tails before their introns are removed. That tends to slow the process, Zhang said, delaying the messenger RNA’s arrival at the ribosome.
How, exactly, that post-transcriptional splicing occurs is an open question that Zhang hopes to answer. It’s not the only one.
“There is no comprehensive study, like our study here, for the animal kingdom,” Zhang said. “That’s why I’m thinking maybe we can apply this idea to it, as well.
“Biologists really want to know about the origin of all plants. But we also want to know: What’s the origin of Homo sapiens? If we can find a good (micro-exon) marker, it can maybe tell us more stories of our history.”
The team, led by Nebraska’s Zhang, Jeffrey Mower and Bin Yu, detailed its findings in the journal Nature Communications, which recognized the paper as an Editors’ Highlight. The trio authored the study with Huihui Yu, Mu Li, Jaspreet Sandhu, Guangchao Sun, James Schnable and Harkamal Walia, all of Nebraska, along with Weibo Xie of Huazhong Agricultural University. The researchers received support from the U.S. National Science Foundation and the Nebraska Soybean Board.